

Introduction: T-cell acute lymphoblastic leukemia (T-ALL) is characterized by proliferation of immature T-cells and accounts for ~15% of pediatric ALL. T-ALL blasts are phenotypically diverse and are sub-classified into pro-, pre-, cortical and mature T-ALL based on the stage of differentiation of the leukemic clone. Early T precursor ALL (ETP-ALL) is a T-ALL subtype associated with higher risk of relapse (Raetz and Teachey, 2016). Bulk sequencing approaches have revealed valuable information about modulated genes in T-ALL; however, little is understood about the interplay between tumor cells and the immune microenvironment. We present a comprehensive single cell RNA sequencing (scRNASeq) analysis of T-ALL samples with the purpose of characterizing the heterogenous tumor and microenvironment cells in order to identify dysregulated genes in leukemia cells and investigate oncogenic signaling pathways.
Methods: We profiled 16,280 cells from 5 diagnostic pediatric T-ALL bone marrow samples using the Chromium single cell transcriptomics platform (10x Genomics, Pleasanton, CA). To compare T-ALL versus healthy bone marrow profiles and identify T-ALL-specific malignant blast cell populations, we included data from 3 healthy pediatric bone marrow samples from a recent study (Caron et al, 2020). Dimension reduction using the Uniform Manifold Approximation and Projection (UMAP) approach was used to identify unique cell type clusters (Becht et al, 2018). Using the TARGET dataset (https://ocg.cancer.gov/programs/target), we further evaluated the prognostic significance of identified malignant blast-specific genes by performing Kaplan-Meier survival analysis and compared gene expression patterns and tumor microenvironment makeup between ETP-ALL and non-ETP T-ALL patient samples.
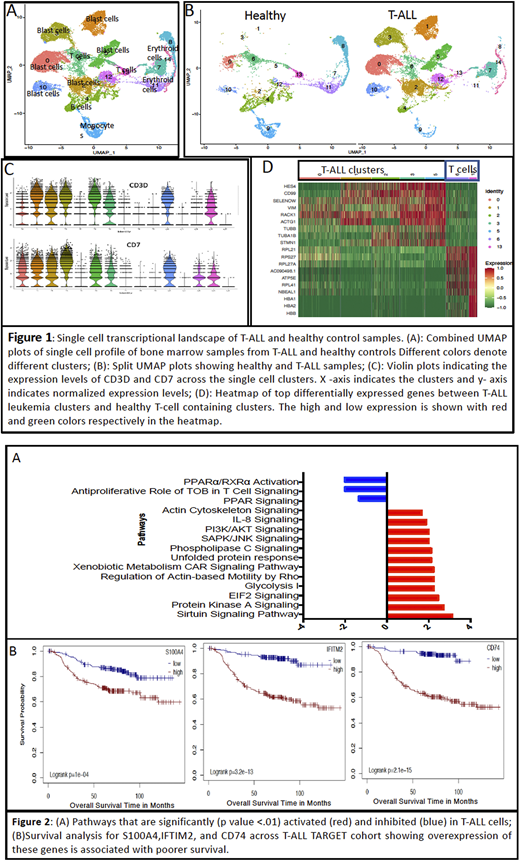
Results: We successfully characterized leukemic blasts (CD7+, CD99+ and CD3D+) and other major immune cell types (T cells, B cells, monocytes, erythroid precursors) using the expression of established marker genes (Fig 1A, C). Clustering analysis revealed patient-specific leukemia blast cell clusters (Fig 1B). Differential expression analysis between CD3D+ patient-specific leukemia clusters and CD3D+ clusters comprised of normal T-cells identified a set of 385 promiscuous genes that are significantly differentially expressed between malignant and normal T-cells (p-value <0.05) despite tumor cell heterogeneity. Among the top genes upregulated in the leukemia clusters are HES4 (a downstream target of NOTCH1), CD99, RACK1, and TUBB (Fig 1D), which have previously been implicated in T-ALL leukemogenesis and chemoresistance (DeDecker et al, 2020; Cox et al, 2016; Lei et all 2016). Further, pathways analysis demonstrated significant activation (Z score >1.3, p-value <0.05) of multiple pathways associated with cancer stemness, cellular growth and proliferation including PI3K/AKT, unfolded protein response, and glycolysis, implicating these genes as oncogenic mediators in T-ALL blasts (Fig 2A). Overexpression of S100A4, IFITM2, and CD74 were significantly associated with poor survival in the TARGET cohort of 241 patients (hazard ratio 2.2, 5.8, and 7.1, respectively, p-value <0.05), indicating their prognostic significance (Fig 2B). We additionally evaluated the expression of our gene set across ETP-ALL and non-ETP T-ALL groups in the TARGET dataset. Multiple leukemic blast-specific genes including ARMH1, CD44, CD74, DNAJC1, and IFITM2 were significantly upregulated in ETP-ALL. Further, deconvolution analysis performed on ETP-ALL vs non-ETP T-ALL samples determined that tumor microenvironment cell types are differentially enriched in these T-ALL groups. We observed enrichment of memory B cells, dendritic cells, NK cells, and M2 macrophages in ETP-ALL samples as compared to non-ETP T-ALL samples. Conversely,lower levels of mast cells and T-regulatory cells were observed in ETP-ALL samples.
Conclusions: We identified a gene signature characterizing heterogenous T-ALL blast populations. External validation using the TARGET dataset identified genes within the signature that are associated with poor outcomes in T-ALL. Differences in the composition of the immune microenvironment in ETP-ALL versus non-ETP T-ALL samples provide a promising area for future study. Further studies will be carried out with relapsed and non-relapsed T-ALL samples to validate this leukemia signature.
Bhasin:Canomiiks Inc: Current equity holder in private company, Other: Co-Founder.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal